- Introduction
- Médicaments contenant des OGM ou consistant en OGM
- Prise de décision
-
Outils pour l'évaluation et la gestion des risques (s'ouvre dans une nouvelle page)
-
Quelques chiffres (s'ouvre dans une nouvelle page)
Introduction
Pour être commercialisé, tout médicament issu des biotechnologies (et donc aussi les médicaments "OGM", c-à-d contenant des OGM ou consistant en OGM) doit faire l'objet d'une autorisation de mise sur le marché délivrée par la Commission européenne sur avis de l'Agence Européenne des Médicaments (EMA). L’accès au marché communautaire pour les médicaments "OGM" est soumis à la procédure centralisée définie dans le règlement (CE) n°726/2004. Si l'autorisation est octroyée, elle est d'emblée valable pour tous les Etats membres de l'Union européenne.
Le demandeur d'une autorisation de mise sur le marché d'un médicament issu des biotechnologies dépose auprès de l’EMA un dossier de demande d'enregistrement qui sera évalué selon des critères scientifiques de qualité, de sécurité et d'efficacité du médicament concerné. Conformément à la procédure centralisée et en fonction du type de médicament, l’évaluation du médicament par l'EMA est faite soit par le Comité des médicaments à usage humain ("Committee for Medicinal Products for Human Use" - CHMP), soit par le Comité des médicaments à usage vétérinaire ("Committee for Medicinal Products for Veterinary Use" - CVMP), soit par le Comité pour les thérapies avancées ("Committee for Advanced Therapy" - CAT).
Pour chaque dossier, l'évaluation est effectuée sous la coordination d'un rapporteur et d'un co-rapporteur désignés par le CHMP, le CVMP ou le CAT. Dans un premier temps, un projet de rapport d'évaluation est préparé, sur base duquel des questions peuvent être envoyées au demandeur. Après les réponses de celui-ci, le rapport d'évaluation final est soumis pour évaluation au Comité correspondant de l'EMA. A l'issue de cette évaluation le CHMP, le CVMP ou le CAT évalue le rapport risque/bénéfice de l'utilisation du produit, et finalise son opinion. Si celle-ci est positive, elle est transmise à la Commission qui adopte la décision finale.
Médicaments contenant des OGM ou consistant en OGM
Les médicaments contenant des OGM ou consistant en OGM constituent un cas réglementaire particulier du fait de dispositions réciproques dans la directive 2001/18/CE (article 12.2) et le règlement (CE) n°726/2004 (articles 6.2 et 6.3). Dans ce cas, la demande doit être accompagnée d'une évaluation des risques pour l'environnement (ERA) conformément aux dispositions de l'annexe II de la directive 2001/18/CE. Elle doit donc contenir le dossier technique complet avec les informations requises par les annexes III et IV de la directive.
L'ERA s’applique aux médicaments contenant des OGM ou consistant en OGM, mais pas aux médicaments produits à partir d’OGM. Parmi ces derniers, on trouve par exemple de l’insuline produite à partir de bactéries recombinantes ou plus récemment, une protéine humaine anticoagulante présente dans du lait de chèvres génétiquement modifiées.
L'ERA est analysée en concertation avec les structures établies par la Communauté ou les États membres conformément à la directive 2001/18/CE. En Belgique, c’est le Conseil consultatif de Biosécurité avec le support scientifique d'experts externes et du SBB, qui procède à cette analyse.
Contributions du Conseil consultatif de Biosécurité et du SBB (voir figure ci-dessous)
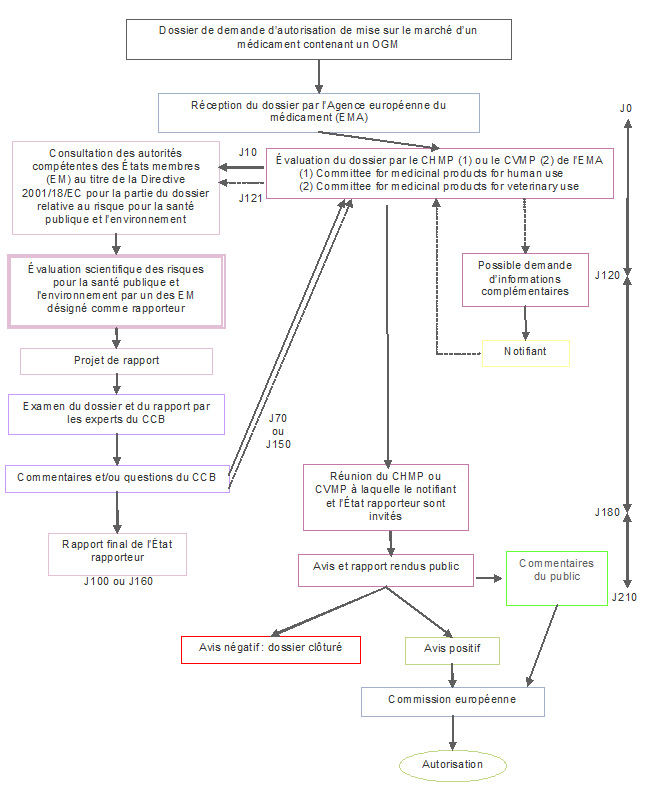
Les projets de rapport d'évaluation préparés par le rapporteur et le co-rapporteur de l’EMA et validés par le CHMP ou le CVMP contiennent une partie dédiée à l’évaluation des risques pour l'environnement (ERA) du médicament "OGM". L’ERA porte sur les risques pour l’environnement biotique, mais également sur les éventuels dangers de l’OGM pour l’entourage de la personne traitée ou pour le personnel soignant, ainsi que les risques de santé publique. Dans cette partie de son rapport, le rapporteur souligne les éventuels manquements dans l’ERA et, si nécessaire, propose une liste de questions à adresser au notifiant.
La partie "ERA" du rapport est communiquée au Conseil consultatif de Biosécurité (CCB) (ainsi qu’à toutes les autorités compétentes désignées dans les autres Etats membres), qui a préalablement reçu accès à la partie "ERA" du dossier.
Le CCB rédige et envoie à l’EMA son avis sur le rapport du rapporteur de l’EMA relatif à la partie "ERA", indiquant éventuellement les points que le rapporteur n’aurait pas pris en compte. L’avis du Conseil indique le cas échéant quelles informations complémentaires devraient être obtenues pour compléter l’évaluation du risque environnemental. Le CCB va ensuite donner un avis à l’EMA pour chaque rapport d’évaluation suivant rédigé par le rapporteur et le co-rapporteur suite aux réponses du notifiant.
Prise de décision
Comme indiqué précédemment, c’est la Commission Européenne qui adopte la décision finale de mise sur le marché pour un médicament soumis selon la procédure centralisée.
Les rapports scientifique complets de chaque médicaments autorisés par la Commission Européenne selon la procédure centralisée suite à une évaluation par le CHMP ou le CVMP de l’EMA sont publiquement disponibles sur le site web de l’EMA (EPAR - European Public Assessment Report). L'EPAR reprend la notice d’information du produit et un résumé de ses caractéristiques traduits dans plusieurs langues européennes, ainsi que la discussion scientifique et les mesures prises pour l'évaluation (en anglais seulement).
Les autorisations de mise sur le marché sont valables cinq ans. Les demandes de renouvellement doivent être introduites à l’EMA au moins six mois avant l’expiration de cette période de validité.